文献阅读(2)scDEAL
标题:Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data
1 亮点
- 它可以使用来自癌症药物敏感性基因组学 (GDSC) 数据库和癌细胞系百科全书 (CCLE)的大量药物反应 RNA-seq 信息来训练并优化模型;
- 为了解释批量和 scRNA-seq 数据之间的数据结构差异,scDEAL 调和了scRNA-seq和bulkRNA-seq的嵌入以确保药物反应标签可从bulkRNA-seq转移到scRNA-seq;
- 为了避免丢失scRNA-seq数据中的异质性,scDEAL在每个训练阶段都包含了用于损失函数正则化的细胞群标签;
- scDEAL 的集成梯度解释推断了药物反应预测的特征基因,从而提高了模型的可解释性。
通过追踪和累积 DTL 模型中每个神经元的积分梯度,进一步确定了被认为直接影响细胞药物敏感性或耐药性的基因特征。
2 scDEAL模型示意图
Overview of scDEAL(single-cell Drug rEsponse AnaLysis)
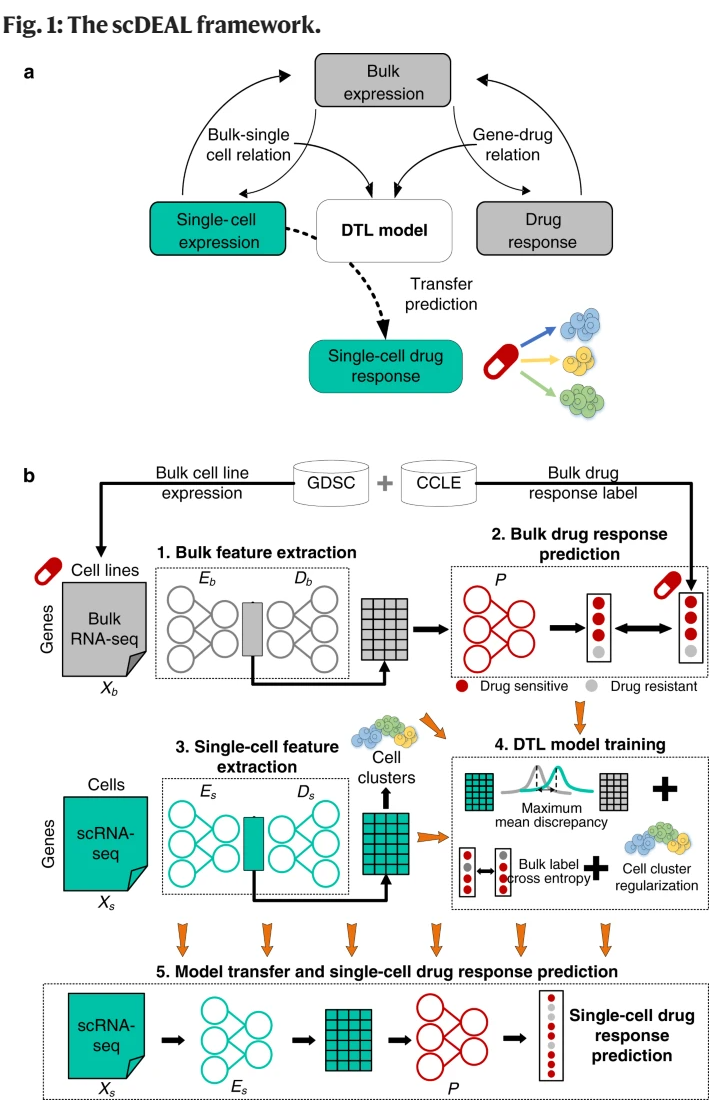 scDEAL示意图
scDEAL示意图 3 实验内容
1 困难
如何在平衡scRNA-seq数据与Bulk RNA-seq数据的同时保持单细胞异质性;
2 解决思路
- 考虑scRNA-seq数据与Bulk RNA-seq数据的噪音特征不同,使用DAE模型引入大量噪音,通过这样的策略迫使scRNA-seq更加接近Bulk RNA-seq数据;
- 通过整合细胞聚类结果来规范 scDEAL 的整体损失函数,从而在训练过程中保留细胞异质性。
评估了由五种药物处理的六个公共 scRNA-seq 数据集的药物反应预测性能。
使用了7个指标:F1分数、 AUCROC、AP 分数、精确度、召回率、调整后的互信息 (AMI) 和调整后的兰德指数 (ARI)
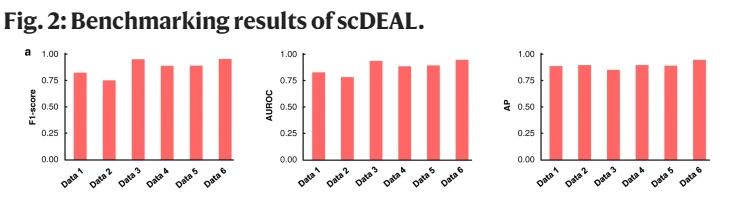 结果图
结果图 3 scDEAL 工作流
整体可以分为两个部分:
- 【step: 1-2】监督学习建立模型预测Bulk RNA-seq标签;
- 【step: 3-5】迁移该模型至scRNA-seq标签预测。
Step 1. Bulk特征提取
通过一个DAE(De-noising Auto Encoder)初步提取Bulk特征,此外这也是Step 3中的预训练模型,DAE 的基本架构由三部分组成:
- 基于原始Xb生成一个噪音矩阵Xb′:
Xb′=B(Xb,pb),
- Encoder(Eb):通过ReLU将Xb′降维至低维表示;
- Decoder(Db):通过低维表示重构Xb′′;
模型概括如下:
minlossrecon(Eb,Db,Xb)=min(MSE(Xb,Xb′′))Xb′′=Db(Eb(Xb′))
Step 2. Bulk药物反应预测
使用MLP在Bulk数据上,训练一个药物反应预测器( Predictor,P ),同时使用Cross Entropy进行参数优化。
模型概括如下:
minlossclass(P,Yb,Yb0)=min(CrossEntropy(Yb,Yb0))Yb=P(Eb(Xb))
Step 3. Single特征提取
同STEP 1,略。
Step 4. DTL模型训练
在这一步是综合训练前三步的模型,需要考虑:两个分布之间的最大平均差异、预测药物反应和真实药物反应之间的交叉熵损失、以及对scRNA-seq数据进行合理聚类的正则项
为了让预测器 P 能够作用于single特征数据,因此需要调和Single特征和Bulk特征。所以采用DaNN模型对 Es 进行调整,引入最大均值差异(MMD)衡量 Es 和 Eb 输出结果的相似性,MMD在该问题中定义如下:
lossMMD(Eb(Xb),Es(Xs))=∣n1i=1∑nϕ(xbi)−m1j=1∑mϕ(xsj)∣H,
此外,在预测器P的训练过程中将两个基因特征之间的相似性加入到分类损失中,以保证 Es 和 Eb 的特征空间具有相似的分布。训练 DaNN 模型以同时更新两个基因提取器( Eb 和 Es )和预测器 P ,模型概括如下:
minlossrecon(Eb,Db,Xb)=min(MSE(Xb,Xb′′))regulizer=CC∑cinCCcosinesimilarity(Xs),
Step 5. 模型迁移以及single特征药物反应预测
经过步骤 4 中训练的 Es 和 P 将被组合用于scRNA-seq 数据中的所有细胞预测单细胞药物反应,通过输入Xs输出连续概率分数Ys,以0-0.5作为耐药细胞,0.5-1作为敏感细胞。
4 预测指标
1 Precision:
Precision=TruepositiveTruepositive+Falsepositive(1)
2 Recall:
Recall=TruepositiveTruepositive+Falsenegative(2)
3 F1-score:
F1−score=TruepositiveTruepositive+0.5∗(Truepositive+Falsenegative)(3)
4 AUROC score:
5 AP score:
AP=i=1∑n(Rn−Rn−1)Pn(4)
6 AMI:
7 ARI:
ARI(P∗,P)=0.5∗[∑i(2Ni)+∑j(2Nj)]−(2N)[∑i(2Ni)∑j(2Nj)]∑i,j(2Nij)−(2N)[∑i(2Ni)∑j(2Nj)],
4 集成梯度法鉴定关键基因(⭐)
应用IG score来表征scDEAL模型中关键输入基因特征。IG 分数表示相对于每个基因表达的梯度积分,作为输入沿着从零表达作为基线到输入表达水平的路径,使用如下所述的黎曼法则对积分进行近似IG score,该方法计算了**输入细胞 x** 的**第i个基因**表达的重要性:
IGi(x)::=(xi−xi′)×∫α=01∂xi∂F(x′+α×(x−x′))d
α,β分别是权重,c是cell,CC是Louvain聚类结果。
这里IG score是通过Python Captum库中的“IntegratedGradients”类进行计算的,要求的输入是【基因表达矩阵】,【训练模型】和【输出标签】,输出结果为与输入表达矩阵形状相同的IG矩阵。因为scDEAL是一个二分类的模型,即有两个输出,因此可以为每个输出获得两个单独的IG矩阵。
5 实验结果
6 参考链接
https://www.nature.com/articles/s41467-022-34277-7open in new window ——原文链接
脚注
